���ӱ����Ƽ���PPI������Ҫȱ�����£���i����������ȶ��ģ�һ����Ҫ�Ʊ��ɳ����Ƽ�������Ӱ����θ�ſ����ʣ���Ҳͬʱ������ҩ������ʱ��Ϊʲô��ʱ��ᷢ�������IJ�ͬ����ii��ҩ������ѧ���ڸ�����죬��Ϊ��Щҩ����Ҫ�ɸ�ҩ���лøCYP2C19��л������ʾ�Ŵ���̬��; ��iii��������ڵ�����ЧӦ���ܳ���24Сʱ����ˣ�PPI���ܳ������ҹ�������;��iv����Щҩ����л�������Ч���ã�ͨ������������ЧӦ�����Եġ������Щ����õ������δ�����ҽ��������٣�������Щ�����ҩ�ォ��������Ϊ��θ����ؼ�����������ѡ���о����֣��ؾ����������ͼ���P-CAB���ķ�չ��ϣ������Ϊ��ѵĽ��������
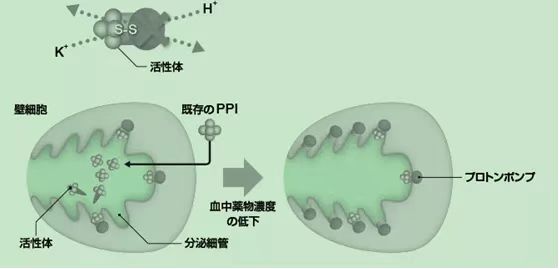
P-CAB����ͨ������H+��K+ -ATPø����PPI�����õIJ�ͬ���ơ�PPIͨ����ø�γɹ��ۼ��������������H+��K+ -ATPø����P-CABͨ����ø��ǻ��K+���λ���ϵ�K+���Ӿ��������������H+��K+ -ATPø����ͼ1��
ͼ1 PPI��TAK-438�����û��ƱȽ�
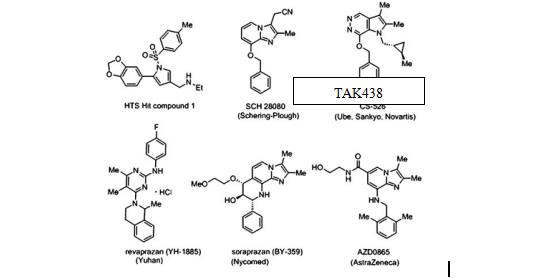
��20����80���������һЩ��ҩ��˾��ͼ����P-CAB�������������ǵĹ�Ч�ζ���[1-2]��ͨ��̽�����о������֡����յ������Ƽ����˷�PPI�ľ����ԣ�ͬʱ��������ǰ������P-CAB���ֳ��IJ����Ч�ʺ�DZ�ڵĸζ��ԡ�
Ϊ�˷���һ���µ������Ƽ����о�����2003���������ø�ͨ��ɸѡ��HTS������������560 000�ֻ�����ɹ�������������������H+��K+-ATPø���ƻ��ԣ��������������¾������õĻ�ѧ�ȶ��ԡ����������������Ϊ��Լ��ԵĽṹ;������λ��1-λ����5-λ�ķ������һ�-����-��������λ����������3-λ�����⣬�����еͷ������ͽṹ�仯��Χ����ŵ㣬���仯ѧ�ṹ�Ƕ��صģ����Ҷ��������Ƽ���ǰ��δ�еġ��о��߽����л�����1��Ϊ�����о���������ΪHTS Hit Compound 1������ṹ��ͼ2��
ͼ2 HTS hit compound 1������������P-CAB��ѧ�ṹ
�о������������л������DZ��������������������˺ϳ��о����������˹�Ч��ϵ��SAR������ͼ3������1��[3]�����ȣ��ϳɻ�����2��֤��������5λ�ϵļ���SAR��������ǽṹ����ʹ�û�������Ե���ά�֡�����������3-λ�ϵ�N-�һ���������ΪN-��������ʱ����������������ӳ���ʮ�������⣬������3��1mg▪kg-1�������ڣ��ڴ���������66��������ڡ���Na+��K+-ATPø��ȣ�������ʾ����H+��K+-ATPø�ĸ�ѡ���ԡ����⣬����ѧ�о�����H+��K+-ATPø�����K+�ľ��������ƣ������û�������P-CAB����ˣ��������ø�Ļ������ӵ�����������������ƣ��Լ��ߵ�øѡ���ԣ��������������ΪP-CAB��һ���������������Ƶ����ú�ѡ�
ͼ3 HTS hit compound 1 ��Ч������
��1 HTS hit compound ��SAR�����
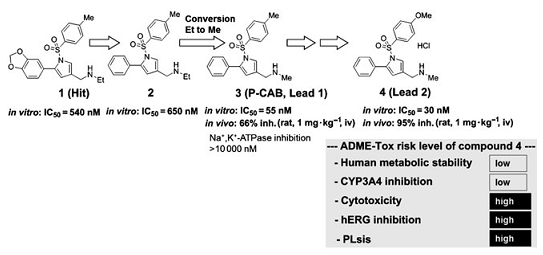
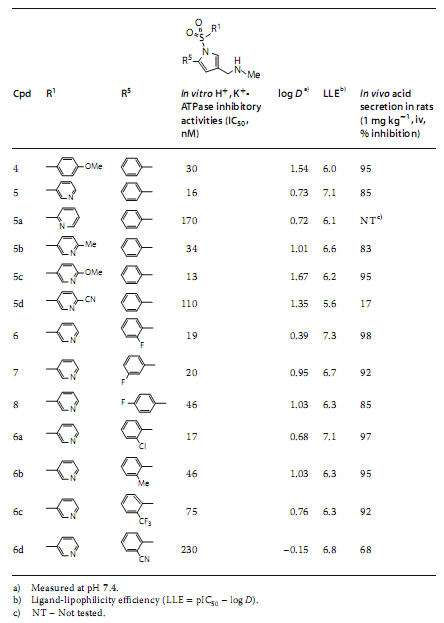
�˺�ѡ����3��Ϊ�ȵ������lead 1�������ۺ��о���SAR����ˣ�N-����������3λ��R3���֣����ֳ��ر���Ч�����ƻ��ԣ���1λ��X��R1���֣��������������ֱ�����ӵķ��㻷��ֱ�����ӵķ�����5λ��R5���֣�������ġ����⣬������2λ��R2���֣���4λ��R4���֣���ȡ����ʱ������û���������ƣ���ͼ3����
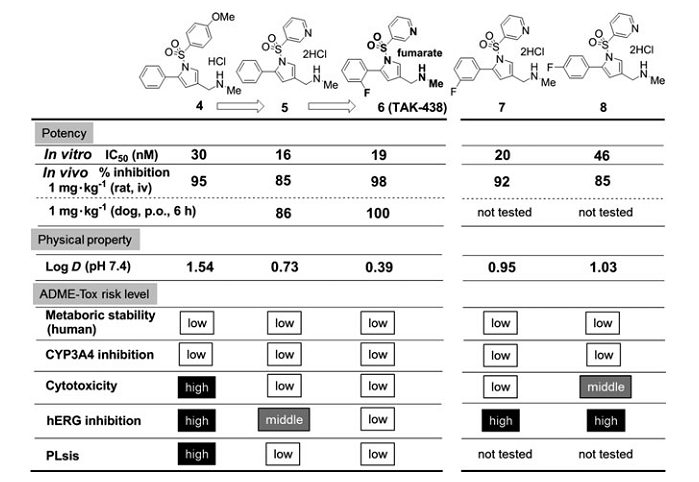
��SAR�����У�Ҳ�������õ�������ѧ���ʺ������л�ȶ��ԵĻ�����4��1mg▪kg-1������ʾ����������90�����ƣ���ǿ�����������ã�95�����ƣ������ܻ�����4��ʾ���м��ֲ�������ADME-Tox���ʣ�����ϸ�����ԣ�hERG���ƺ����ߵ���֬����PLS�����գ������Ա���Ϊһ��DZ�ڵ��ȵ������
�����µĺ�ѡ��������Ż�����ֱ�Ӻ���ȷ�����ڻ����lead 2���ȵ��Ż�һֱ�ڳ������С�ADME-Tox���ݵļ��ͨ�����Խϵͻ��ԵĻ�������С�����ʵ�ʲ�����log Dֵ������ϸ����������֮�����������������ԡ����⣬���۲쵽������log Dֵ��hERG����֮���������ԣ������ͼ4��
ͼ4 ����������IJⶨlog Dֵ�����ⶾ������֮��������
������Щ���֣��о����ܹ��γ�һ�ּ��裬��ͨ������������logDֵ�������������������������ADME-Tox�ֲ��������������ش����⡣һ�������Ǻϳɷ������������뼫�Ի��Ž�����������1-λ����Ҫ������ϵ͵Ļ�ѧ��Ӧ�ԡ���һ�������ǵ�log D����������ڻ����������͡�����һ��ʹ��15-��-5��Ϊ���Ӽ����ºϳɷ��������ǰ�����⣬�Ӷ���������������1λ������ּ��Ի��š��о��߳���ʹ�ýṹ��������Ч��/���ⶾ������֮��Ĺ�ϵ�����һ���⣬��������������1λ�ϵļ��Ի��ŵ�λ�ã�Ȼ������������5λ������ȷ����3-��ऻ���Ϊ���ȡ����֮һ�������log Dֵ[4]��
�������ŵ������TAK-438����Ϊ��ѡҩ��Ľṹ
����ƵĻ����������������֬��Ч�ʣ�LLE = pIC50-logD��ֵ��Ϊҩ�������Ե�ָ�꣬�������2��
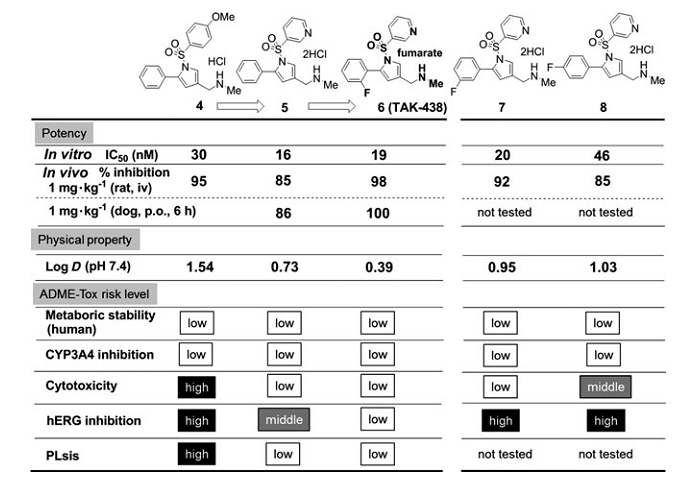
ͼ5 ͨ���Ż�ѡ����6��TAK-438���������ŵ������
��2��lead compound 4�Ż�����������
��Ϊ���۸��ֺϳɻ�����Ľ������3-��ऻ�����R1���ֵõ�������5����ͼ5��������ʾ0.73�ĵ�log Dֵ�����������ϸ�����Ժ���֬�����ķ��գ���û��������������Ч�ʡ����⣬���н�2-F-Ph��������R5���ֵĻ�����6�����ϵ͵�log DֵΪ0.39��ͬʱ����ǿ��H+��K+-ATPø���ƻ��ԡ����⣬���ڴ�������ʾ��ǿ��Ч�������ڹ�����ʾ�������Ŀڷ����ԡ�������6��������֬��Ч��LLE����ֵ�ϸߣ���ֵΪ7.3������hERG���Ƶķ���Ҳ��͡���ˣ��û������ʵ��ؼ�С������ADME-Tox�����Ĺ�ע��
��Ϊ�Աȣ����в�ͬλ�õ�Fԭ�ӵĻ�����7��8��ʾ�Ȼ�����5���ߵ�logDֵ��Ȼ������Щ����������Ч�ʺ�ADME-Tox���ʷ������Բ��㡣
��ˣ��о���ѡ����6��TAK-438���������ŵ��������Ϊ�����ĺ�ѡ�������ͼ5����
�ں����������У�TAK-438�������ٴ�ǰ�о��У�ͬ��Ҳ���ٴ������гɹ���ȡ���������Ľ�������ٴ��о��У�TAK-438�������õİ�ȫ�Ժ����õ������ԡ�
1��������Ҫ�ο�J��nos Fischer and Wayne E.Childers��д���鼮Successful Drug Discovery Volume 2�еĵ�10����Discovery of Vonoprazan Fumarate (TAK-438) as a Novel, Potent and Long-Lasting Potassium-Competitive Acid Blocker��
2�������½��������߸��˶�����ҩ������һЩ�뷨�����⣻��ӭ��λ��ʦ����ָ����
1.Parsons, M.E. and Keeling D.J. (2005) Novel approaches to the pharmacological blockade of gastric acid secretion. Exp. Opin. Investig. Drugs, 14, 411�C421.
2.Kahrilas, P.J., Dent, J. and Lauritsen, K.(2007) A randomized, comparative study of three doses of AZD0865 and esomeprazole for healing of refluxl esophagitis. Clin. Gastroenterol. Hepatol.,5, 1385�C1391.
3.Nishida, H., Hasuoka, A., Arikawa, Y. et al. (2012) Discovery, synthesis, and biological evaluation of novel pyrrole derivatives as highly selective potassium-competitive acid blockers. Bioorg. Med. Chem., 20, 3925�C3938.
4.Arikawa, Y., Nishida, H., Kurasawa, O. et al. (2012) Discovery of a novel pyrrole derivative 1-[5-(2-fluorophenyl)-1-(pyridin-3-ylsulfonyl)-1H-pyrrol-3-yl]-N-methylmethanamine fumarate (TAK-438) as a Potassium-Competitive Acid Blocker (P-CAB). J. Med. Chem., 55, 4446�C4456.
����Ǣ̸��������
��ϵ��ʽ��13635699896����ͬ�ţ�
����Ǣ̸��������
��ϵ��ʽ��15156091013����ͬ�ţ�
��ӭ����ȫ�����صĺ������ݰ����˾�ι۽�����



 ������� 34012302000810��
������� 34012302000810��